Практика инспектирования по GMP

Перспективы сближения национальных подходов, взаимного использования и признания результатов

В 1969 г. ВОЗ предложила государствм-членам заключить соглашение глобального уровня об использовании Системы удостоверения качества лексредств в международной торговле [1]. Система базировалась на взаимном признании результатов инспектирования производителей по правилам GMP.
Сегодня, без малого 50 лет спустя, около 100 стран осуществляют зарубежные инспекции по GMP. В результате компании-экспортеры, в т.ч. имеющие адекватные системы качества, могут подвергаться многократным обследованиям в течение одного года со стороны национальных и различных зарубежных регуляторов. Это ведёт к дублированию усилий, т.е. к непроизводительным затратам надзорных органов и промышленности.
Сложившаяся ситуация объясняется рядом причин. Очевидно, помимо соображений национального суверенитета, существенную роль играет недостаток взаимного доверия между регуляторными органами различных стран. Последнее может быть связано с различиями, фактическими или предполагаемыми, в части опыта и ресурсов национальных инспекторатов. Наряду с этим в силу различий в нормативно-правовой базе и в стандартах GMP результаты проверок зарубежных инспекторов могут рассматриваться как не вполне отвечающие национальным требованиям. В ряде случаев специфические формы оплаты труда инспекторов могут препятствовать взаимному признанию результатов обследований и сокращению объема собственных проверок.
Расширение и углубление сотрудничества между регуляторами, учет и признание результатов проверок заслуживающих доверие инспекторатов могут содействовать оптимизации использования ресурсов.
Для этого необходимо дальнейшее сближение на глобальном уровне практики инспектирования и соответствующей нормативно-правовой базы между странами. В качестве первого шага в этом направлении предлагается собрать методом опроса информацию о процедурах инспектирования в различных странах. Такой проект можно рассматривать как дополнение к ежегодным опросам, проводимым Федерацией европейских инновационных фармацевтических производителей (EFPIA) [2].
Развитие практики инспектирования по GMP: краткий исторический обзор
В настоящее время правила GMP в значительной степени гармонизированы на глобальном уровне. Руководства по GMP Евросоюза и PIC/S, практически идентичные, признают примерно 50 стран-участниц этих организаций. К этому числу можно добавить ряд государств, образующих субрегиональные инициативы экономической интеграции, такие как Евразийский экономический союз, АСЕАН, Совет по сотрудничеству стран Персидского залива.
Кроме того, многие страны, не входящие в указанные объединения, признают и используют в той или иной степени Руководство ВОЗ по GMP, незначительно отличающееся от стандартов Евросоюза и PIC/S. Это касается государств с растущим фармацевтическим производством и экспортным потенциалом, в частности Китая, Индии, Вьетнама, Индонезии, Египта, Пакистана и ряда других стран.
Следует отметить, что документы ICH: Q7, Q9 и Q10 содействуют сближению различных нормативов по GMP, не гармонизированных между собой, в частности требований FDA США, правил ЕС, АСЕАН и ВОЗ. При этом, однако, глобальное сближение инструментов оценки соответствия требованиям GMP, т.е. процедур инспектирования, отстает от гармонизации самих нормативов.
Фармацевтические инспекции, т.е. официальный надзор за деятельностью аптек, аптечных лабораторий и складов, производителей лексредств и предприятий оптовой торговли ими существовали во многих странах задолго до наступления эпохи GMP. Однако в современном значении этот термин стал использоваться лишь вслед за внедрением правил GMP. В дальнейшем его применение стало распространяться на область GDP.
Первоначально национальные процедуры инспектирования по GMP различались в связи с различиями в традициях, законодательной базе и имевшихся ресурсах. В 60-х годах прошлого века были инициированы усилия по гармонизации практики инспектирования, вначале на субрегиональном уровне (Скандинавские страны). Этот процесс продолжился затем на региональном/межрегиональном (Европейское экономическое сообщество, PIC) и глобальном (ВОЗ) уровнях. В результате в последующих десятилетиях был достигнут широкий международный консенсус в отношении общих принципов организации и проведения инспекций в целях контроля соблюдения правил GMP.
Тезис о важной роли фармацевтической инспекции в общем процессе регулирования лекарственного рынка впервые был сформулирован ВОЗ в 1990 г. В руководстве о принципах функционирования регуляторных органов [3] отмечалось, что ведущим элементом любой системы контроля за обращением фармацевтических препаратов является их регистрация. При этом, однако, уточнялось, что без фармацевтического инспектората, поддержанного контрольно-аналитической лабораторией, невозможно обеспечить соблюдение условий регистрации ¹.
¹ В этой связи следует отметить, что функции инспектората могут быть значительно шире, нежели контроль за соблюдением правил GMP на производстве. В ряде стран инспекторы готовили проекты национальных правил GMP и активно участвовали в программах обучения производственного персонала работе по этим правилам. Кроме того, группы самоинспектирования предприятий отрасли, университетские программы подготовки кадров и консалтинговые структуры частного сектора нередко опираются на опыт официальных инспекторов по GMP.
В последующие годы ВОЗ выпустила Руководство по инспектированию фармацевтических производителей [4], в котором нашли отражение важные элементы этого вида надзорной деятельности. Отмечались, в частности, следующие положения:
- Двойная цель государственного инспектората по GMP – контролировать соблюдение общих стандартов GMP и проверять соответствие условий производства, включая контроль качества, конкретных препаратов требованиям соответствующих
регистрационных документов. - Первейшая обязанность инспектора – представить детальный отчет о фактическом соблюдении стандартов производства.
- Существуют различные виды инспекций: плановая, предрегистрационная, сокращённая, последующая, оценка системы качества и др. Инспекции могут проводиться как с предварительным уведомлением производителя, так и без оного.
- Типичные частота и продолжительность инспекций: один раз в два года, от нескольких дней до двух недель.
- Порядок планирования и проведения инспекций.
- Вступительное и заключительное совещания с руководством предприятия.
- Последовательность осмотра предприятия.
- Использование вопросников.
- Доступ инспекторов к документации по самоинспектированию или по аудитам качества.
- Отбор образцов для анализа.
- Структура отчета об инспектировании.
- Последующие регуляторные действия.
Дополнительные руководящие материалы ВОЗ касались инспектирования производства исследуемых препаратов, предрегистрационных обследований, систем качества национальных инспекторатов, формата отчетов об инспектировании и сертификатов GMP [5-8].
Наряду с этим PIC-PIC/S и Евросоюз сформулировали ряд специфических рекомендаций, имеющих практическую важность для национальных инспекционных служб [9-19]. Первая версия руководства ЕС Проведение инспекций фармацевтических производителей или импортеров было выпущено в 1996 г. Текущий вариант этого норматива включен в сборник Еврокомиссии и Европейского агентства по медикаментам в отношении союзных процедур по инспектированию и обмену информацией [20].
В результате совместных действий ВОЗ, PIC/S и Евросоюза в 90-х годах прошлого столетия были согласованы на международном уровне следующие общие принципы функционирования инспекционных служб:
- Цель инспекции – оценить производство готовых лекарственных препаратов на соответствие стандартам GMP. В тот период инспектирование производителей АФИ осуществляли лишь отдельные страны.
- Важнейшим результатом обследования площадки является отчет об инспектировании, не сертификат GMP.
- Инспектирование тесно связано с другими регуляторными функциями: с регистрацией препаратов, с контролем качества образцов, отобранных на рынке и др.
- Данную функцию исполняет специализированная административная структура: инспекторат по GMP.
- Инспекторат в составе службы охраны общественного здоровья страны. В этой связи следует выделить положение, зафиксированное в Конвенции PIC (1970 г.): «Инспекции проводятся национальными органами здравоохранения» [9](выделено для целей настоящей статьи). Как правило, инспекторат является частью регуляторного органа по лексредствам.
- Инспектирование по GMP носит официальный статус в отличие от действий, проводимых по принципу «услуга, выполняемая по инициативе заявителя». Этот статус подтверждается обязательным характером проверок, а также требованиями устранения выявленных нарушений (САРА по современной терминологии). Также в соответствии с официальным статусом инспекций к нарушителям в необходимых случаях применялись регуляторные санкции. Расходы на проведение инспекций чаще всего оплачивались из бюджета регуляторного органа.
Что касается импортируемых фармацевтических препаратов, США, Австралия, отдельные государствачлены ЕС проводили прямое инспектирование зарубежных производителей. Однако, с учётом требуемых в этих случаях значительных затрат, а также нередких технических осложнений, в качестве наиболее перспективной формы контроля предприятий-экспортеров было выделено заключение двух- и многосторонних соглашений между странами о взаимном признании результатов инспектирования. Альтернативный, легко реализуемый подход заключается в принятии правовых актов в отношении одностороннего признания результатов инспектирования специалистами отдельных, конкретно определенных стран. Примером служит решение Колумбии
признавать результаты американских инспекторов. Аналогичные примеры имеются и в Европе.
В 1970 г. ряд европейских государств подписали Конвенцию о взаимном признании инспекций в отношении производителей фармацевтических препаратов (PIC) [9]. Страны-участницы приняли и признали инспекции фармацевтических производителей, выполненные уполномоченными органами других стран-участниц эквивалентными их собственным национальным инспекциям. В середине 90-х годов этот же принцип взаимного признания инспекций фармацевтических производителей был принят Европейским
сообществом (в настоящее время Европейский союз).
К этому моменту число странучастниц Конвенции выросло, и она приобрела межрегиональный масштаб. Однако фактически она перестала функционировать, в связи с чем было подписано альтернативное соглашение: Схема сотрудничества по фармацевтическим инспекциям (PIC/S). В рамках этого механизма обмен отчетами об инспекциях и сведений о постинспекционных действиях компаний не приводил автоматически к сокращению числа обследований ².
² Косвенно Схема сотрудничества содействовала ограничению программ зарубежного инспектирования, поскольку создавала основу заключения двухсторонних соглашений о взаимном признании инспекций.
Подобная информация использовалась в целях подготовки к плановым инспекциям. В настоящее время в Схеме сотрудничества участвует свыше 50 государств, считая страны, подавшие заявки на вступление и находящиеся на этапе подготовки к вступлению. Это четверть числа стран-членов ООН.
Практически одновременно с созданием PIC в 1969 г. ВОЗ рекомендовала странам-экспортерам и импортерам лексредств участвовать в Системе удостоверения (сертификации) качества фармацевтических препаратов в международной торговле [1]. Целью Системы было содействовать международной торговле медикаментами в глобальном масштабе путем взаимного признания результатов инспекций. При этом, однако, не было предусмотрено мер независимой внешней оценки регуляторных возможностей странучастниц. По этой причине выданные в рамках Системы сертификаты не получили широкого признания со стороны импортеров. В Схеме участвуют свыше 140 стран. Это соглашение используется для обмена информацией об импортируемых готовых препаратах и АФИ. Очевидно, что возможности этого механизма сотрудничества недоиспользованы.
В настоящее время отсутствуют соглашения глобального уровня о взаимном признании результатов инспектирования по GMP. Недавно Европейское агентство по медикаментам (ЕМА) опубликовало обзор международных инициатив по регулированию оборота лексредств [21]. Согласно этому документу ни одна программа глобального международного сотрудничества не содержит в своих планах сближения практики инспектирования по GMP.
Возникновение альтернативных подходов
Вслед за появлением в 1987 г. международных стандартов ИСО по системам качества (семейство ИСО 9000) инспектораты по GMP стали использовать новые подходы. В этой связи необходимо уточнить, что сертификация на соответствие этим стандартам, хотя и преследует те же цели, что и инспектирование по GMP (оценка возможностей производителя в сфере обеспечения качества продукции), отличается существенным образом от этого вида деятельности. Поскольку сертификация по ИСО 9000 не является регуляторной функцией, она не носит обязательного характера, осуществляется структурами честного сектора, по инициативе заявителя в момент, когда последний готов к обследованию. Проверка нацелена на конечный результат: сертификат, не на отчет, отражающий состояние дел. Сертификат соответствия не увязан с качеством конкретных товаров, он не согласован с документами типа досье на продукт, отражающими технологию и другие аспекты производства тех или иных изделий.
При выдаче сертификата благоприобретателем является заявитель: он получает рыночное преимущество перед конкурентами. В практическом плане подобную сертификацию нельзя считать нацеленной преимущественно на защиту общественных интересов.
Наиболее важным элементом, заимствованным из практики сертификации по ИСО 9000 является внутренняя система качества инспектората. По этой теме имеются инструктивные материалы международных организаций в форме итоговых документов семинаров PIC/S (1994 и 1998 гг.), руководств ВОЗ и Евросоюза (2002 и 2003 гг. соответственно). Согласно этим документам система качества инспектората по GMP имеет целью предотвращение и недопущение конфликта интересов персонала. Порядок получения оплаты за проведение инспекций не должен влиять ненадлежащим образом на процесс и результаты инспектирования. Независимость и непредвзятость инспекторов должна быть обеспечена строгим соблюдением инструкций, процедур, правил и кодексов поведения. В материалах ВОЗ указано также, что инспекторы не должны быть под контролем фармацевтических производителей, они должны проходить процедуры аттестации и лицензирования. Кроме того, заимствованными из ИСО являются концепции Показателей Качества (Quality Metrics), Непрерывного Усовершенствования (Continuous Improvement) и Статистического Контроля процессов. Данные концепции уже используются в Правилах надлежащей Производственной Практики ЕС и США.
Наряду с этим ВОЗ рекомендует, чтобы каждый отчет об инспекции рассматривался в порядке, предусмотренном системой качества инспектората. Это позволяет руководству оценивать отчеты, подготовленные отдельным инспектором или их небольшой группой, по возможности с привлечением наиболее опытных коллег. При этом руководитель проверяет соблюдение соответствующих инструкций в ходе инспектирования, соответствие рекомендаций изложенному состоянию дел и в целом степень объективности отчета. В результате повышается качество отчетов, заключения более обоснованы, а рекомендации лучше проработаны.
Со временем в работу регуляторов фармацевтического сектора стали проникать и другие элементы из практики сертификации по ИСО 9000. Примерами являются обследование в виде услуги по заявкам производителей, получение оплаты от инспектируемых предприятий, акцент на сертификат и пониженное внимание к отчетам, включая указания об устранении выявленных нарушений. Аналогичные тенденции наблюдались и в работе международных организаций. Это касается программы предквалификации ВОЗ, практики выдачи сертификатов соответствия Европейской фармакопеи в отношении АФИ, порядка инспектирования производителей субстанций регуляторами стран ЕС.
Современные тенденции в практике инспектирования по GMP
В результате развитие практики инспектирования по GMP пошло по необычному пути: в начале от национальных различий к признанию общих принципов, а затем в противоположном направлении: к использованию альтернативных подходов, в целом
негармонизированных. В настоящее время практика инспектирования в отдельных странах и регионах мира сближена лишь частично. Национальные процедуры различаются в ряде важных аспектов. Ниже перечислены некоторые примеры различий.
- Положение инспектората в регуляторной системе.
- Выполняемые функции, помимо инспектирования по GMP (другие виды инспектирования, лицензирование производителей, дистрибьюторов и др.).
- Уровень нагрузки, измеряемый числом держателей производственных лицензий на одного инспектора.
- Методы финансирования функции инспектирования.
- Подход к инспектированию как к услуге, выполняемой по заявке производителя.
- Инспектирование без предварительного уведомления.
- Подходы к инспектированию производителей АФИ и зарубежных площадок.
- Использование удаленных (т.н. «бумажных») инспекций.
- Использование вопросников в процессе выездных обследований площадок.
- Доступность отчетов о самоинспектировании или о внутренних аудитах качества для официальных инспекторов.
- Формы доведения до предприятия письменной информации о результатах инспектирования.
- Взаимоотношения инспекционной службы с регистрационным органом, с контрольно-аналитическими
лабораториями. - Рассмотрение отчетов об инспектировании в инспекторате.
- Использование инспекторатом методов управления рисками для качества в соответствии с рекомендациями ICH (Q9), PIC/S и ВОЗ.
- Признание сертификатов GMP, выданных регуляторными органами, применяющими строгие требования.
- Использование сертификатов формата, рекомендованного ВОЗ(СРР).
Практика инспектирования в качестве услуги, выполняемой по заявке производителя, и связанная с этим оплата затрат за счет проверяемых предприятий заслуживают дополнительных замечаний. Очевидным преимуществом такого подхода является финансовая независимость инспектората от госбюджета. Следует вместе с тем учитывать, что в рамках такого подхода затраты, формально оплачиваемые промышленностью, фактически отражаются на стоимости препаратов и, следовательно, ложатся на потребителя – пациента.
К тому же в определенных условиях работники фармацевтического сектора и общественность могут рассматривать выдачу сертификатов как коммерческую деятельность, что негативно скажется на доверии к этой функции. В этой связи можно вспомнить о разразившемся в 2001 г. скандале по поводу сертификации по ИСО 9000 ³.
³ 30 ноября 2001 г. генсек ИСО д-р Лоуренс Ейшер заявил: «Мы регулярно получаем жалобы относительно сертификатов,
необоснованно выданных компаниям, не прошедшим должного аудита». (Источник: пресс-релиз ИСО № 805).
Заслуживает упоминания ещё один момент, обычно ускользающий от внимания. Оплата расходов по инспектированию самими предприятиями препятствует международному сотрудничеству в этой сфере. Если взносы проверяемых компаний являются единственным или основным источником финансирования инспектората, то предложения об обмене отчетами и о сокращении на этой основе числа обследований будут противоречить экономическим интересам инспекторов. Именно этой причиной, скорее всего, объясняется отсутствие сотрудничества в области сертификации по ИСО 9000.
Целесообразность дальнейшего сближения практики инспектирования
Достигнутый уровень согласования порядка работы и, соответственно, степень взаимного доверия инспекторатов обеспечивают функционирование ряда международных соглашений, а также позволяет использовать элементы гибкости в национальном законодательстве и регуляторных требованиях отдельных стран. Так, результаты инспекций, выполненных уполномоченным органом страны-члена Евросоюза, принимаются соответствующими органами всех других государств, входящих в ЕС, этот же принцип действует и в других регионах мира, например в рамках АСЕАН. Кроме того, Евросоюз имеет соглашения о взаимном признании результатов с Австралией, Канадой, Израилем, Новой Зеландией, Швейцарией и Японией. Австралия заключила двухсторонние соглашения с Канадой, Японией и Сингапуром. Предполагалось, что аналогичные договоренности будут действовать между государствами Евразийского экономического союза (Россия, Беларусь, Казахстан, Армения и Кыргызстан). Следует отметить, что в настоящее время практика взаимного признания результатов инспекций между государствами Евразийского экономического союза еще не внедрена.
Тем не менее, преимущества взаимного доверия и признания сертификатов GMP не используются в полной мере. Одна из причин – недостаток доверия между национальными инспекторатами, что может быть связано с различиями в возможностях или в законодательстве и процедурах.
Несколько лет назад секция промышленной фармации FIP сравнила степень требовательности инспекторатов ряда стран при проведении обследований. Приведенные ниже результаты говорят о существовании национальных различий в этой сфере.
Тщательность инспектирования по GMP (в убывающем порядке)
данные М. Анисфельда, председателя секции промышленной фармации FIP
- США, Великобритания
- Скандинавские страны, Австралия, Канада
- Государства Южной Европы, Венгрия, Япония
- Бразилия, Саудовская Аравия
Следует отметить, что за последние пять лет произошли сдвиги в гармонизации многих инспекторатов. Главными препятствиями к полной эквивалентности являются различия в возможностях национальных инспекторатов и практическая трактовка правил надлежащей производственной практики.
В последние годы существенные изменения произошли в Азии. Помимо отмеченной выше Японии Ю. Корея и Тайвань проводят зарубежные инспекции адекватного уровня, уделяя должное внимание деталям. Китай начал инспектировать поставщиков лексредств за пределами своей страны, однако на данный момент при этом предъявляются не очень жесткие требования, очевидно, в связи с ограниченными ресурсами и опытом работы надзорных органов.
В 1999 г. было заключено торговое соглашение между Евросоюзом и США, включавшее раздел о взаимном признании результатов инспектирования по GMP. Этот раздел не вступил в силу в связи с тем, что, по мнению американской стороны, результаты проведенных совместных инспекций указывали на недостаточно высокий уровень инспектирования в некоторых странах ЕС. В настоящее время завершается цикл совместных инспекций, охватывающий все государства-члены Евросоюза. Готовится новое соглашение; этот проект имеет политическую поддержку как в США, так и в ЕС [22].
Некоторые страны с укрепляющимися регуляторными системами склонны бессистемно копировать или приспосабливать организационные подходы государств, предъявляющих строгие требования к операторам отрасли. В других случаях, например, в некоторых новых независимых государствах, используются необычные административные формы, как-то создание разных структур для инспектирования локальных и зарубежных производителей. В этой связи можно отметить, что ВОЗ не рекомендует копировать регуляторные решения других стран без учета общей ситуации и мировых тенденций.
Недостаток доверия к инспекторатам других стран ведёт к повторным обследованиям производителей (до 9 и более раз в год в отношении одной площадки), что означает дублирование усилий и дополнительные затраты как регуляторов, так и промышленности. Как уже отмечено выше, расходы предприятий, так или иначе, переводятся на потребителей. Кроме того, многочисленные инспекции задерживают доступ пациентов к необходимым лексредствам, поскольку сертификаты GMP часто требуются в составе регистрационных документов.
Международная гармонизация усилий в этой сфере продолжается. Так, например, в последнее время был пересмотрен формат отчетов об инспектировании; это относится к рекомендациям ВОЗ [23]. В целом пакет руководств ВОЗ в отношении фармацевтических инспекций весьма внушителен. Он, однако, состоит из разрозненных документов и не полон. Составляющие его материалы были опубликованы в период между 1990 и 2016 годами; некоторые из них нуждаются в пересмотре. С учетом глобальных тенденций и последних изменений в мировой практике представляются желательными дополнительные усилия. Возможно, имеет смысл актуализировать рекомендации ВОЗ в этой области и объединить разрозненные документы в единое руководство.
Ситуация для подобных действий на глобальном уровне выглядит благоприятной. В ходе состоявшейся недавно 17-ой Международной конференции органов, регулирующих оборот лексредств (ICDRA), Кейптаун, Ю. Африка, 27 ноября — 2 декабря 2016 г. обсуждались возможности более тесного сотрудничества на глобальном уровне между различными инициативами по гармонизации. В феврале 2016 г. вступил в силу пересмотренный меморандум о взаимопонимании между ВОЗ и PIC/S. Укрепляется взаимодействие между PIC/S и Евросоюзом, включая Европейское агентство по медикаментам и руководителей регуляторных органов стран ЕС. Объединяются усилия PIC/S, ICH и Международной коалиции регуляторных органов (ICMRA). Обсуждается возможность участия инспекционных подразделений партнерских организаций в PIC/S.
Исследование EFPIA
Европейская федерация ассоциаций фармацевтической промышленности (EFPIA) объединяет инновационные фирмы стран ЕС и их зарубежные филиалы, в т.ч. за пределами Европы. Начиная с 2003 года Федерация проводит ежегодные исследования, касающиеся инспектирования своих членов. Согласно данным, полученным от примерно 660 производственных площадок, расположенных по всему миру, за последние 12 лет зарубежные инспекции проводили 64 страны. Здесь и ниже государства-члены ЕС считаются за одну страну. Это означает, что фактическое число стран, осуществляющих обследования европейских фирм с учетом их филиалов в других регионах мира, приближается к 100.
В период с 2011 по 2015 гг. наибольшее число зарубежных инспекций выполнялось странами со строгими регуляторными требованиями: США (90) и государствами ЕС (65). На втором месте группа стран с укрепляющимися контрольно-надзорными системами: Ю. Корея, Бразилия, Турция, Япония и Кения. Страны этой группы осуществляли от 20 до 60 зарубежных инспекций каждая. К третьей группе отнесены страны с регуляторными органами, имеющими ограниченный опыт; на каждую из них приходится от одной до 20 инспекций.
Интересно отметить, что в последние годы страны первых двух групп сокращают число своих зарубежных инспекций, тогда как в третьей группе, напротив, отмечается активизация инспекционных программ. Китай, например, провел 5 зарубежных инспекций в 2011 г. и 16 в 2015 г. У Беларуси этот показатель вырос вчетверо: с трёх до 12 за те же годы.
В 2015 г. 640 производственных площадок прошли 1495 обследований со стороны национальных и иностранных инспекторатов. В зарубежных инспекциях участвовали 34 страны. Наиболее активны в этом плане были США, государства ЕС, Ю. Корея, Бразилия, Турция, Япония и Кения. Почти половина (46%) инспекций выполнили инспектораты-члены PIC/S в странах, также участвующих в Схеме сотрудничества по фарминспекциям. Наряду с этим значительное число обследований провели государства, имеющие ограниченные возможности в этом плане, такие как Кения, Ливия, Уганда, Нигерия и Судан.
Из 1495 инспекций 167 (11%) были предрегистрационными. В это число входили 71 национальная и 96 иностранных. Предрегистрационные проверки осуществляли страны с различным уровнем участия в международном сотрудничестве: члены PIC/S (Канада, Япония, США, страны ЕС, Ю. Корея), кандидаты на вступление в PIC/S (Беларусь, Бразилия, Казахстан, Мексика), а также не имеющие официальных контактов с PIC/S (Китай, Кения).
Из 640 площадок, данные по которым вошли в обзор EFPIA за 2015 г., 5 прошли по 15 инспекций каждая. 52 площадки приняли по 4 и более инспекций, 570 площадок сообщили о прохождении от одной до 4 проверок и 118 не были обследованы в этом году.
Менее 10% обследований были проведены без предварительного уведомления предприятия. Свыше 95% инспекций дали положительные результаты, т.е. не повлекли за собой регуляторных действий. Лишь в 0,1% случаев были выявлены нарушения, приведшие к задержкам поставок препаратов.
Средняя продолжительность зарубежных инспекций составляла 4 дня, что на один день меньше в сравнении с локальными проверками. Чаще всего в обследованиях участвовали по два инспектора. Ресурсы, выраженные в человеко-днях на одну инспекцию, зависели от национального инспектората. Например, для Тайваня, Мексики, Китая, Канады, Колумбии, Саудовской Аравии этот показатель составлял 10 и более человеко-дней. Для Турции, Бразилии, Беларуси, Ю. Кореи, стран ЕС, Японии и Казахстана – от 5 до 10 человеко-дней. Менее 5 человеко-дней отмечалось в отношении Нигерии, Уганды, Австралии, России и Кении.
Во многих случаях компании обязаны представить документацию до начала инспекций. Имеющиеся данные показывают, что инспектируемые компании тратят в 10 раз больше средств, чем регуляторы на подготовку, прохождение и постинспекционные действия. Связанные с этим затраты промышленности оцениваются в среднем в 70 человекодней на одну инспекцию. По данным за 2015 г. суммарные ресурсы, вложенные членами EFPIA в процедуры проведения инспекций, составили 70 тыс. человекодней. Средний размер пошлины или сбора на проведение одной инспекции составлял 30 тыс. евро. За 2015 г. на инспекции фирм-членов EFPIA регуляторы потратили более 75 000 человекодней; фирмы потратили более €80 млн на поддержку инспекций.
Ниже приведены выводы и рекомендации, сформулированные Федерацией по материалам своих обзоров. Представляется, что эти соображения заслуживают внимания профильных международных организаций и общественности.
Фармацевтическая промышленность всё больше становится глобальной. В производстве одного лекарственного препарата может участвовать значительное число площадок (от 6 до 30), расположенных в различных странах. В ряде случаев в регистрационные документы включается более одной площадки с целью уменьшения рисков прекращения производства и связанной с этим нехваткой препаратов на рынках. Одна площадка может иметь несколько лицензий на производство, что вызывает необходимость повторных инспекций.
В результате ни один регуляторный орган не в состоянии проинспектировать все производственные площадки, участвующие в цепочке фармацевтического снабжения.
С учетом изложенного, регуляторам следует стремиться к признанию и использованию, по возможности, данных других контрольно-надзорных органов на принципах анализа рисков и экономии имеющихся ресурсов.
В последние годы число зарубежных инспекций стабилизировалось на высоком уровне. Несмотря на усилия по сотрудничеству национальных и региональных регуляторных органов, существует значительное дублирование в сфере надзора за производственными площадками. Позиция промышленности сводится к тому, что многочисленные повторные инспекции не всегда основаны на анализе рисков для качества.
Инспекции требуют значительных ресурсов как от регуляторов, так и от промышленности. В ряде случаев уровень сборов на эти цели мало оправдан. Добавленная стоимость функции инспектирования в целом не высока. Результаты обзоров указывают на возможность оптимизации использования выделяемых на эти цели ресурсов.
Имеющиеся данные говорят о том, что общепризнанная в глобальном масштабе система сертификации по GMP могла бы принести положительные результаты. Она содействовала бы сокращению дублирования усилий в сфере инспектирования. Это, в свою очередь, позволило бы перераспределить ресурсы в направлении сегментов цепочки фармацевтического снабжения, в наибольшей степени связанных с рисками для качества.
С тем, чтобы облегчить взаимное признание результатов инспекций и сократить число повторных обследований по GMP, необходимо дальнейшее сближение процедур инспектирования и принципов организации уполномоченных органов в различных странах и экономиках [2, 24-26].
В этой связи целесообразно привести слайд, представленный д-ром С. Роннингером, фирма Амджен, г. Зуг, Швейцария на 17-ой Международной конференции органов, регулирующих оборот лексредств (ICDRA). Слайд указывает на важный фактор: учёт и использование данных, полученных от других инспекторатов, может содействовать лучшему использованию ресурсов при одновременном увеличении числа площадок, в отношении которых имеется достаточная информация.
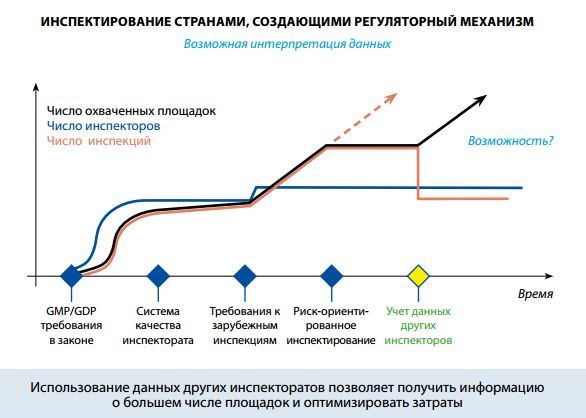
Обзорные данные EFPIA дают общее представление о ситуации в сфере инспектирования по GMP, рассматриваемой с позиций промышленности. Эти данные, однако, будучи сфокусированы на статистике частоты инспектирования и оценке используемых ресурсов, не охватывают всех аспектов проблемы, имеющих практический интерес.
С учетом этого желательно подумать о структурированном подходе к сопоставлению практики инспектирования и выявлению положительного опыта в этой области на уровне стран под руководством компетентной профильной структуры глобального уровня. Это могла бы быть Программа основных лексредств ВОЗ (ЕМР). Значительным опытом в этой области располагает PIC/S; с учетом этого, а также принимая во внимание растущее число стран-участниц этой организации, рекомендации PIC/S в части укрепления инспекционных служб могли бы принести существенную пользу.
В качестве первого шага в этом направлении предлагается провести исследование в форме опроса в отношении национальных процедур инспектирования. Его можно рассматривать как вклад в усилия по глобальной гармонизации в сфере регулирования фармацевтического производства. Результаты такого проекта по сбору фактических данных, в случае успеха, могут быть распространены в форме публикации или информационного материала (доклада). Они также могут лечь в основу рабочего документа ВОЗ, направленного на обновление регуляторных материалов.
Предложенный проект мог бы дополнить ежегодные обзоры EFPIA. Он планируется в форме одномоментного сбора информации в отношении инспекционной практики отдельных стран и, следовательно, будет касаться как инновационных компаний, так и производителей дженериков. Вопросы для сбора информации адресованы не предприятиям, но физическим лицам. Это специалисты с опытом работы в промышленности, в регуляторных органах, в образовательных программах или в консалтинговых структурах частного сектора.
Источники информации:
- Resolution adopted by the Twenty-Second World Health Assembly. WHA22.50. Quality Control of Drugs.
- S. Rönninger, J. Berberich, V. Davoust, P. Kitz, A. Pfenninger, Landscape of GMP/GDP inspections in research-based pharmaceutical industry – Part I: Data, Pharm. Tech. Europe, 2017, in press; Part II: Opportunities, Pharm. Tech. Europe, 2017, in press.
- Guiding principles for small national drug regulatory authorities. 1990. WHO TRS 790.
- «Provisional guidelines on the inspection of pharmaceutical manufacturers». 1992. WHO TRS 823, Annex 2.
- Pre-approval inspections. 2002. WHO TRS 902, Annex 7.
- Quality systems requirements for national good manufacturing practice inspectorates. 2002. WHO TRS 902, Annex 8.
- Model certifcate of good manufacturing practices. 2003. WHO TRS 908, Annex 5.
- Guidance on good manufacturing practices: inspection report. 2003. WHO TRS 908, Annex 6.
- Convention for the Mutual Recognition of Inspections in Respect of the Manufacture of Pharmaceutical Products. Geneva – October 1970.
- The Role of Inspection and Testing in Relation to the Marketing Authorization. The collected papers presented at a Seminar held in Louvain-la-Neuve from 15 to 17 September 1993. Published by the Secretariat to the Pharmaceutical Inspection Convention (PIC) and Pharmaceutical Inspection Co-operation Scheme (PIC/S).
- Manufacture and Inspection of Active Pharmaceutical Ingredients. The collected papers presented at a Seminar held in Naantali from 10 to 12 June 1997. Published by the Secretariat to the Pharmaceutical Inspection Convention (PIC) and Pharmaceutical Inspection Co-operation Scheme (PIC/S).
- Non-technical aspects of inspection. The collected papers from Civil Service College presented at a Seminar held in Oxford (United Kingdom) from 8 to 10 September 1999. Published by the Secretariat to the Pharmaceutical Inspection Convention (PIC) and Pharmaceutical Inspection Co-operation Scheme (PIC/S).
- The Inspection of Products Derived from Biotechnologies. The collected papers from the French Agency for the Safety of Health Products (AFSSAPS) presented at a Seminar held in Colmar (France) from 25 to 27 October 2000. Published by the Secretariat to the Pharmaceutical Inspection Convention (PIC) and Pharmaceutical Inspection Co-operation Scheme (PIC/S).
- Recommendation on Quality System Requirements for Pharmaceutical Inspectorates. Pharmaceutical Inspection Convention. Pharmaceutical Inspection Co-operation Scheme. PI 002-3. 25 September 2007.
- Aide-memoire. Inspection of pharmaceutical quality control laboratories. Pharmaceutical Inspection Convention. Pharmaceutical Inspection Co-operation Scheme. PI 023–2 25 September 2007.
- Aide-memoire. Inspection of utilities. Pharmaceutical Inspection Convention. Pharmaceutical Inspection Co-operation Scheme. PI 009-3 25 September 2007.
- Aide-memoire. Inspection of active pharmaceutical ingredients. Pharmaceutical Inspection Convention. Pharmaceutical Inspection Cooperation Scheme. PI 030-1 13 January 2009.
- A recommended model for risk-based inspection planning in the GMP environment. Pharmaceutical Inspection Convention. Pharmaceutical Inspection Cooperation Scheme. PI 037-1. 1 January 2012.
- PIC/S Assessment & Joint Reassessment Programme. Revised PIC/S audit checklist based on Evaluation Guide for GMP Regulatory Compliance Programme (by Health Canada). Pharmaceutical Inspection Convention. Pharmaceutical Inspection Co-operation Scheme. PS/W 1/2005 (Rev. 2) Annex 20 October 2014.
- Compilation of Community Procedures on Inspections and Exchange of Information. European Commission Health & Consumer Protection DirectorateGeneral. European Medicines Agency. EMA/572454/2014 Rev 17. 3 October 2014.
- Connecting the dots. Towards global knowledge of the international medicine regulatory landscape: mapping of international initiatives. EMA 2016.
- Dara Corrigan, The Mutual Reliance Initiative: A New Path for Pharmaceutical Inspections in Europe and Beyond.
- Guidance on good manufacturing practices: inspection report. 2016. WHO TRS 996, Annex 4.
- Peter J. Kitz. Opportunities for International Collaborations in inspections. FDA/PQRI Conference Sept 16 and 17, 2014.
- EFPIA Position Paper, Final, v8a – 19/05/2014. Enhanced Good Manufacturing and Good Distribution Practices (GMP/GDP) Inspection Efciency.
- EFPIA. Annual Regulatory GMP/GDP) Inspection Survey 2015 Data.
- Stephan Rönninger, Amgen (Europe) GmbH. EFPIA. The GMP/GDP Inspection Landscape and opportunities for better use of resources: Industry view. 17th International Conference of Drug Regulatory Authorities (ICDRA), Cape Town, S. Africa, 27 November-2 December 2016.
Благодарность:
Авторы выражают благодарность следующим специалистам, внесшим вклад в подготовку статьи:
- Michael Anisfeld, фирма Глобфарм, г. Дирфилд, США,
- Alan Chalmers, компания Фарма Интернейшнл, г. Алшвил, Швейцария,
- Tor Graberg, компания АстраЗенека, г. Сёдертальё, Швеция,
- Elizabeth Meyers, компания Амджен, г. Зуг, Швейцария
Источник: http://gmpnews.ru/2017/04/praktika-inspektirovaniya-po-gmp/