Статья посвящена Приложение 11 к Руководству GMP ЕС "Компьютеризированный системы" в редакции от 14 января 2011 года, вступившей в действие 30 июня 2011 года.
Тимур Кабадаи,Управляющий партнер компании CONVALgroup (Турция)
Н.В. Дынька, вице-президент группы компаний ВИАЛЕК
Сегодня невозможно представить деятельность успешной, развивающейся компании без эффективного использования информационных технологий. Сферы применения компьютеризированных систем постоянно расширяются и неуклонно движутся вперед семимильными шагами. Сейчас применяемые компьютеризированные системы являются не только зеркалом соблюдения принципов GMP, но и фактором конкурентоспособности предприятия. Есть множество примеров используемых компьютеризируемых систем – это и ЛИМС, и пакеты поддержания электронного досье серии (например, на базе 1С: Производство), и автоматизация общего документооборота, системы управления зданиями и сооружениями, системы мониторинга чистых помещений, инженерных систем и т.п.
Тенденции последних лет по стремительной автоматизации производства уже ни у кого не вызывают сомнений в том, что именно требования Приложения 11 GMP будут задавать тон всех регуляторных инспекций ближайших десятилетий.
Впервые 11-ое приложение GMP ЕС [1] было принято в 1997 году. На протяжении 14-ти лет его требования воспринимались специалистами по-разному. Одни их «демонизировали», другие и всерьез не воспринимали. Все возрастающий уровень использования компьютеризированных систем и их повышенная сложность, инициативы FDA в отношении новых подходов к валидации процессов [3], концепции РАТ [4] и инициативы ИСО по внедрению систем информационной безопасности потребовали пересмотра Приложения 11 GMP.
В целом, новая версия Приложения 11 является последовательным шагом в обновлении GMP с учетом проактивного применения методологии управления рисками.
Революционным положением новой версии является то, что все используемые компьютерные приложения должны пройти валидацию, а работоспособность IT-инфраструктуры необходимо подтвердить квалификацией (в ГОСТ Р 52249-2009 использован термин «аттестация»). Обратите внимание на эти 10 слов – впервые в регуляторной практике введено прямое требование о необходимости проведения квалификации IT-инфраструктуры, т.е. программно-аппаратных средств, которые обеспечивают функционирование основного компьютерного приложения. От того, насколько качественно и удобно организована IT-инфраструктура, в той или иной степени зависит успешное протекание всех бизнес-процессов предприятия.
IT-инфраструктура включает в себя следующие звенья:
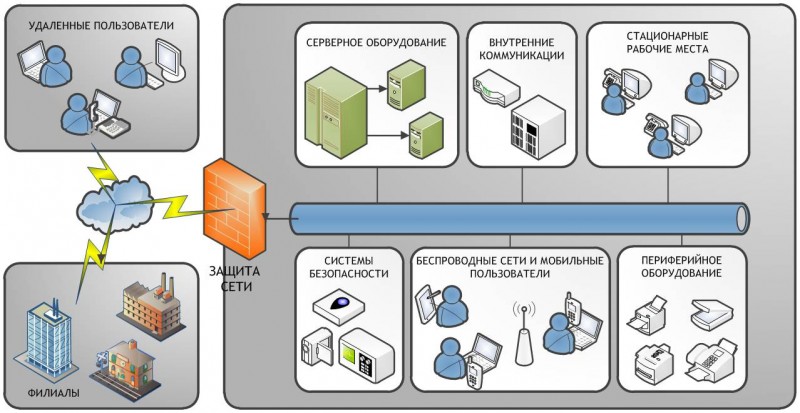
Схема IT-инфраструктуры. Компьютеризированная система включает ряд компонентов программно-аппаратных средств, которые вместе выполняют определенные функции
Область применения Приложения 11 GMP
11-е Приложение GMP распространяется на все формы компьютеризированных систем, которые используются на предприятии в сфере регулирования GMP. Это относится и к электронным таблицам (например, Excel, Visual Basic), и к базам данных собственной разработки.
Жизненный цикл системы
Под «жизненным циклом системы» понимается период времени, который включает все фазы жизни системы, начиная с момента принятия решения о ее создании и заканчивая моментом ее полного изъятия из эксплуатации у пользователя [5]. Фазы жизненного цикла включают проектирование, разработку спецификации, программирование, проведение тестирования, монтаж, эксплуатацию и техническое обслуживание системы.
По сути, жизненный цикл системы наглядно демонстрирует принцип циклического спиралевидного развития. Жизненный цикл системы повторяется, причём каждое повторение этого цикла приводит к более глубокому пониманию процессов обработки данных. Реально концепция новой системы возникает уже в процессе внедрения и начале эксплуатации разрабатываемой системы. По окончании очередного цикла система обновляется. Длительность цикла обновления системы определяется конкретными потребностями производства, а также техническим прогрессом и развитием идеологии обработки данных.
В настоящее время в России действующим стандартом, который предусматривает стадии и этапы создания системы, является ГОСТ 34.601-90 [6]. Однако в этом стандарте многие процессы отражены недостаточно, а некоторые положения уже значительно устарели.
Основой мировой практики являются международный стандарт ISO/IEC 15288:2008 [7], описывающий жизненный цикл компьютеризированной системы и стандарт ISO/IEC 12207:2008 [8] соответственно описывающий жизненный цикл программного обеспечения. Оба стандарта могут совместно использоваться при закупке и установке компьютеризированной системы с необходимым программным обеспечением.
Ниже представлены основные требования 11-го Приложения GMP ЕС в заявленной последовательности, рекомендации Европейского медицинского агентства (ЕМА) [1, 2] и некоторые комментарии от авторов данной статьи.
Структура обновленного приложения 11 GMP
Принцип
Общие положения
1 – Управление рисками
2 – Персонал
3 – Поставщики компонентов и услуг
Фазы проекта
4 – Валидация
Эксплуатация системы
5 – Данные
6 – Проверка точности
7 – Хранение данны
8 – Вывод данных на печат
9 – Контрольные журнал
10 – Управление изменениями и конфигурацией
11 – Периодическая оценк
12 – Безопасность
13 – Управление происшествиями (инцидентами)
14 – Электронная подпись
15 – Выпуск серии
16 – Бесперебойная работа
17 – Архивирование
Глоссарий (словарь)
Новый текст 11-го приложения GMP декларирует принцип «не навреди», а именно: «Замена ручных операций какой-либо компьютеризированной системой не должно приводить к снижению качества продукции, контроля процесса или достигнутого уровня обеспечения качества. Компьютеризированная система не должна увеличивать совокупный риск для функционирующего процесса». Очевидно, что заявленный принцип является своеобразной «перестраховкой» – ведь, по своей сути, применение компьютеризированной системы способствует значительному улучшению управляемости процесса и уровня обеспечения качества, так как практически устраняется человеческий фактор, а соответственно и риски ошибок персонала. Есть и другая сторона, устраняя одни риски, мы невольно можем существенно повысить статус других. Именно поэтому в новом тексте 11-го Приложения GMP особое внимание уделено управлению рисками.
1 – Управление рисками
Управление рисками должно осуществляться на протяжении всего периода эксплуатации компьютеризированной системы. Все решения об объеме валидации и степень усилий по управлению достоверностью данных должны основываться на обоснованной и документально подтвержденной оценке рисков такой системы.
11-ое приложение GMP предполагает, что управление рисками будет применяться при оценке критичности тех или иных ее компонентов, оценки степени ее влияния на качество и безопасность продукции, на достоверность собираемых данных. Разработка спецификации на систему (URS), формирование рабочих инструкций для персонала, система защиты и мероприятия по обеспечению бесперебойной работы должны учитывать результаты документированной оценки рисков.
Реализация программы управления рисками для компьютеризированной системы подразумевает идентификацию всех возможных ее отказов, ошибок персонала, потери данных и т.п., оценку вероятности их наступления и степени критичности, исходя из возможных последствий для потребителя. И соответственно выполнение комплекса мероприятий, направленных на устранение все значимых рисков.
2 – Персонал
Для каждой системы должен быть назначен «владелец системы» – это один из руководителей предприятия, ответственный за доступность, работоспособность, обслуживание компьютеризированной системы и безопасность имеющихся данных. Одним из основополагающих принципов надлежащей эксплуатации системы является тесное взаимодействие IT-специалистов с ключевым персоналом предприятия, а именно, владельцем самой системы, владельцами соответствующих бизнес-процессов и Уполномоченными Лицами. Важно обеспечить условия, при которых все задействованные лица будут обладать достаточной компетентностью, уровнем доступа и четким распределением обязанностей.
3 – Поставщики компонентов и услуг
Поддержание компьютеризированных систем требует выполнения большого объема работ – подбора, обеспечения, монтажа (инсталляции), конфигурирования, интеграции, валидации и технического обслуживания (например, через удаленный доступ), изменений и обработки данных. Учитывая повышенную сложность компьютеризированной системы, часто приходится привлекать специалистов третьей стороны, т.е. лиц, не находящихся в прямом подчинении владельца лицензии на производство или импорт продукции. Поэтому для гарантий качества компьютеризированной системы крайне важно поддерживать систему управления поставщиками компонентов и связанных с ней услуг.
Ключевыми факторами выбора продукта или поставщика услуг являются компетентность и надежность поставщика. Для проверки выполнения всех требований пользователей необходимо обеспечить оценку документации на все приобретаемые стандартные коммерческие продукты с привлечением авторизованных пользователей. Все подписываемые контракты должны включать четкие формулировки обязательств третьей стороны. Аналогичный подход должен использоваться и для организации работы IТ-подразделения предприятия.
В свою очередь, при проведении инспекции на соответствие GMP инспектор имеет право запрашивать результаты оценки разработчика и поставщика программного продукта или системы. При этом решение о целесообразности аудита поставщика рекомендуется принимать на основании оценки рисков.
Фазы проекта
Любая компьютеризированная система должна быть валидирована. Руководству предприятия необходимо признать, что валидация системы – это не одноразовое событие. Она должна начинаться со стадии определения требований пользователя (уже при составлении URS) и заканчиваться внесением последней записи о том, что система снята с эксплуатации. Существует множество предубеждений, связанных с этим вопросом. Например, «Мы покупаем уже валидированную систему», «Наша система уже пару лет используется, ни разу не было сбоев – это и есть валидация», «Мы протестировали программное обеспечение, значит мы провели валидацию», «Валидация нужна только для новых систем» и т.п. Но это только предубеждения!
Валидация системы должна быть проведена при ее запуске, для уже функционирующих системы должна быть проведена ретроспективная валидация, и периодически необходимо проводить ревалидацию. Периодичность ревалидации необходимо устанавливать исходя из критичности самой системы для качества выпускаемой продукции или сохранности накапливаемых данных.
Документация по валидации должна охватывать все этапы жизненного цикла системы. Дополнительно в нее необходимо включить все записи по управлению изменениями и любым отклонениям, выявленным при валидации.
Необходимо поддерживать актуальный перечень всех значимых систем и набор (опись) их функциональных возможностей в отношении GMP. Все критические системы должны иметь: текущее описание системы (ее архитектуру), уточняющие физические и логические схемы, алгоритмы обработки данных и зоны взаимодействия (интерфейс) с другими системами и процессами, аппаратурное и программное компьютерное обеспечение, а также меры обеспечения безопасности.
Требования пользователей должны прослеживаться по всему жизненному циклу системы. Спецификации пользователя (URS) должны описывать необходимые функции системы, основываться на принципах GMP и результатах документированной оценки рисков. Производители должны быть в состоянии подтвердить свои стандарты, протоколы, критерии соответствия, процедуры и записи, основанные на их оценке риска. Авторизованные пользователи должны предпринять все рациональные меры для получения гарантий того, что система разработана в соответствии с приемлемой системой управления качеством.
Валидация заказных систем или систем, модифицированных под требования заказчика, должна включать официальную оценку и отчетность по качеству и работоспособности каждого из этапов жизненного цикла системы. Нужно представить доказательства соответствующих методов и сценариев испытаний. В частности, должны учитываться ограничения системного (процессного) параметра, защиты памяти и обработки ошибок. Для компьютеризированных инструментов и режимов испытаний необходимо обеспечить документированную оценку их достоверности.
Учитывая последующий рост базы данных, испытания при валидации необходимо проводить «под нагрузкой».
При возможности переноса данных в другой формат или в другую базу данных в объем валидационных испытаний нужно включать проверки, подтверждающие неизменность данных при таком переносе. Соответствующие предостережения должны выполняться при переносе данных в конце жизненного цикла системы.
А на частый вопрос «Зачем разрабатывать спецификацию пользователя (URS) на уже давно используемую систему?» имеется простой ответ – ретроспективная валидация, т.е. подтверждение возможности использования старой системы по ее прямому назначению с учетом принципов GMP, включает gap-анализ (анализ просчетов, т.е. сопоставление текущей и желаемой ситуации) и оценку соответствия системы требованиям ее пользователей. Без документально оформленной URS сделать это невозможно. Вообще-то без URS невозможна любая валидация.
5 – Данные
Для устранения рисков, связанных с потерей или искажением данных, системы, которые могут осуществлять электронный обмен данными с другими системами, должны быть оснащены встроенными контрольными устройствами правильного и безопасного ввода и обработки данных.
6 – Проверка точности
Все критические данные, которые вводятся вручную, должны проходить дополнительную проверку точности. Такая проверка может выполняться вторым оператором или проверенными электронными средствами. С помощью инструментов управления рисками необходимо установить критичность и возможные последствия неправильно или неточно введенных в систему данных.
Для электронных таблиц (например, таблицы Excel, Visual Basic) с расчетами должна исключаться возможность ошибочного ввода данных несоответствующего типа (например, текст в числовое поле), случайного перезаписывания или удаления формул и т.п. Все используемые электронные таблицы должны пройти верификацию – документальную проверку точности формул, считываемости данных и всех типов применяемых алгоритмов.
7 – Хранение данных
Необходимо обеспечить защиту данных с помощью физических и электронных устройств. Данные, находящиеся на хранении следует проверять на доступность, считываемость и правильность. Данные должны быть доступны на протяжении всего периода хранения, который не может отличаться от периода хранения данных на бумажных носителях.
Должен быть организован архив версий шаблонов электронных таблиц (для возможности идентификации ошибок).
Регулярно должно проводиться резервное копирование всех важных данных. Целостность и правильность резервных данных, а также возможность их восстановления следует проверять при валидации и периодически оценивать.
Необходимо получить гарантии того, что протоколы электронных подписей будут действительны на протяжении всего периода хранения.
8 – Вывод данных на печать
Необходимо обеспечить возможность получения четких печатных копий данных, которые хранятся в электронном виде. Для записей, поддерживающих выпуск серии, следует обеспечить возможность формирования распечаток, отображающих наличие каких-либо изменений с момента внесения первичной записи.
Часто такая функция не предусмотрена поставщиком программного обеспечения, поэтому с первого дня эксплуатации системы необходимо поддерживать альтернативные способы – например, распечатку и заверение первичных записей до выдачи разрешения на реализацию серии, или контрольную распечатку при самоинспекциях или внешних инспекциях и сличение с сопутствующими данными на бумажных носителях и т.п.
9 – Контрольные журналы
По результатам оценки рисков рассматривается возможность оснащения системы функцией регистрации всех значимых для GMP изменений и удалений, а также их причин (т.н. генерируемый системой «контрольный журнал»). Контрольные журналы должны быть доступны, поддерживаться в понятной для всех форме и регулярно проверятся.
10 – Управление изменениями и конфигурацией
Любые изменения в компьютеризированной системе, включая ее конфигурацию, должны вноситься только управляемым способом в соответствии с одобренной процедурой.
11 – Периодическая оценка
Для подтверждения рабочего состояния систем и их соответствия принципам GMP необходимо проводить периодическую оценку. Проверка должна включать оценку текущего перечня функциональных характеристик, записи по отклонениям, происшествиям, проблемы, историю обновлений, отчеты о производительности, надежности и безопасности, а также все валидационные отчеты.
12 – Безопасность
Для обеспечения санкционированного доступа к системе требуется физический и/или логический контроль на соответствующем уровне. К приемлемым способам защиты от несанкционированного доступа относятся использование ключей, идентификационных пластиковых карточек, персональных кодов с паролями, система биометрической идентификации, ограниченный доступ к компьютерному оборудованию и зонам хранения данных. Приемлемый уровень безопасности определяется степенью критичности компьютеризированной системы для качества выпускаемой продукции.
Надлежит регистрировать все операции по получению, изменению и блокировке доступа. Должна существовать система управления данными и документацией, позволяющая идентифицировать пользователя, который вводит, изменяет, подтверждает или удаляет данные, включая регистрацию даты и времени проведения таких манипуляций.
В последние годы для обеспечения информационной безопасности активно применяется международный стандарт ISO/IEC 27001 [9].
13 – Управление происшествиями (инцидентами)
Необходимо регистрировать и анализировать любые происшествия, а не только системные сбои и ошибки данных. Для каждого критического происшествия следует установить основную причину и выполнить достаточные корректирующие и предупреждающие действия, направленные на ее устранение.
14 – Электронная подпись
Электронные протоколы могут подписываться в электронном виде. При этом на предприятии электронные подписи (8 апреля текущего года вступил в силу Федеральный закон Российской Федерации от 6 апреля 2011 г. № 63-ФЗ «Об электронной подписи») должны иметь такой же статус, как и рукописные подписи, быть постоянно связанными со своим протоколом (записью) и отражать время и дату ее использования.
15 – Выпуск серии
Компьютеризированная система может использоваться для регистрации и выдачи разрешений на выпуск серий продукции (в соответствии с 16-м приложением GMP). Допуск к выдаче разрешений в такой системе может быть только у Уполномоченного Лица. При этом система должна четко идентифицировать и регистрировать работника, выдающего разрешение на выпуск конкретной серии. Для этой цели используется электронная подпись.
16 – Бесперебойная работа
Важно обеспечивать бесперебойную работу компьютеризированных систем. Для каждой системы, поддерживающей на предприятии критические процессы, необходимо предусмотреть комплекс альтернативных мер, направленных на постоянное поддержание процессов при ее отказе (например, оперативное использование ручной или альтернативной системы). Для оценки времени, достаточного для принятия таких действий, необходимо основываться на риске и учитывать реалии системы и бизнес-процесса. Необходимо надлежащим образом зарегистрировать и протестировать результативность таких действий.
17 – Архивирование
Все данные, которые архивируются в системе, должны проверяться на доступность, считываемость и правильность. Необходимо обеспечить и проверять возможность поиска и извлечения таких данных на случай значимых изменений в системе (например, замену компонентов компьютера или программ).
Особенности для мелких изделий
К так называемым «мелким изделиям» (small device) относится часто используемое настольное оборудование. Примерами такого оборудования являются спектрофотометры, ВЭЖХ, лабораторные весы и др. Жизненный цикл таких изделий, как правило, полностью находится под надзором разработчика. Часто такие изделия комплектуются микропроцессорами и встроенным программным обеспечением. Доступ пользователя к настройкам их параметров ограничен. Собираемые данные хранятся не постоянно и не могут быть изменены пользователем. Соответственно, связанные с такими изделиями риски незначительны.
Задача предприятия-пользователя – адекватно определить способности разработчика к развитию программного обеспечения под требования пользователей в соответствии с едиными стандартами качества.
Но и в таких ситуациях GMP-инспектор может потребовать:
Также необходимо учитывать требования ИСО 10012 [10] к средствам измерений, регулировки таких приборов должны быть защищены пломбами. Верификация, проводимая при начале эксплуатации прибора, может рассматриваться как PQ (Performance Qualification).
На радость специалистов по информационным технологиям 11-ое Приложение GMP является не единственным нормативным документом для организации управления компьютеризированными системами. Известны стандарты ISO, FDA, TPD, IEEE, GAMP и другие.
Законодателем моды в области компьютерной инженерии, биомедицинских технологий и телекоммуникаций принято считать профессиональное общество IEEE (Institute of Electrical and Electronics Engineers), в состав которого входит свыше 350 000 специалистов из более, чем 150 стран мира. IEEE создает 30% всей публикуемой в мире литературы по электроинженерной и компьютерной тематике.
Руководство по GAMP (Good Automated Manufacturing Practice) [11], изданное ISPE (International Society for Pharmaceutical Engineering), направлено на оказание помощи поставщикам компьютеризированных систем в обеспечении им надлежащей функциональности и документального оформления в соответствии с требованиями GMP. Обзор текущей версии GAMP мы планируем представить читателям в последующих выпусках журнала.
Литература
Опубликовано в журнале «Чистые помещения и технологические среды» №2, 2011 г. www.cleanrooms.ru
Новости
|
||||||||||||||||||||||||||
 |
Вебинар: Комплексные решения Testo для фармацевтической отрасли Приглашаем принять участие в предствоящем вебинаре "Комплексные решения Testo для фармацевтической отрасли". |
 |
Вебинар: Логгеры-регистраторы параметров микроклимата в фармацевтической отрасли Приглашаем принять участие в предствоящем вебинаре "Логгеры-регистраторы параметров микроклимата в фармацевтической отрасли". |
 |
Вебинар: Измерительные приборы Testo для фармацевтической отрасли В ближайший четверг, 18 ноября приглашаем вас на бесплатный вебинар «Измерительные приборы Testo для фармацевтической отрасли». |
|
22 мар |
Мировой уровень покрытия GMP составляет порядка 60%
|
|---|
|
19 фев |
Эксперт: Маркировка лекарств может обойтись среднему предприятию в 6 млн евро
|
|---|
|
12 окт |
Сергей Цыб о проверках и повышении качества российских лекарств
|
|---|
|
31 авг |
Разработан проект Руководства по обеспечению целостности данных
|
|---|
|
18 май |
Правила GDP. Квалификация транспортных средств
|
|---|




